Predicting Blood Tests Results Using The Gut Microbiome
Your gut bacteria tell more than you think about you
It is well known that the human gut Microbiome stores a huge amount of valuable medical information about each and every one of us.
From metabolic diseases indicators to food sensitivities and even autism and depression, the bacteria composition in our gut serves as our unique fingerprint that secretly hides many of our strengths and weaknesses.
What is our Gut Microbiome?
Your body is full of trillions of bacteria, viruses and fungi. They are collectively known as the microbiome.
Specifically, your gut Microbiome refers to all bacteria living in your intestines. In fact, there are more bacterial cells in your body than human cells, which basically means we are more bacteria than humans.
While some bacteria are associated with diseases, others are actually extremely important for your immune system, heart, weight and many other aspects of health.
Predicting Blood Tests
At DayTwo, we collect and analyze thousands of gut microbiome samples as part of our core product. In our research group, among other research initiatives, we focus on finding new links between the bacteria living in our gut and various health conditions.
As part of our efforts to find such links (aka biomarkers), we were able to predict a few blood tests results using only the gut microbiome. This reinforces the fact that the microbiome has a significant role in our health, and just by looking at it and mapping the bacteria in it, we can produce lots of interesting insights.
Our Training Process
We used 30,000 users for which we have their microbiome samples together with their HbA1C, Fasting Glucose and Fasting Triglycerides values.
We trained 3 machine learning models, one for each blood test, and we used 3,500 microbiome taxonomic features I.E. each feature represents the abundance of a specific bacteria species in the microbiome.
For the modeling part, we used LGBM models running with 5-fold cross-validation and tuned with bayesian hyperparameter optimization.
Features Explainability
One of the requirements when analyzing bioinformatics data in general, and specifically with biomarkers discovery models, is the ability to explain the decisions made by the model and the possibility of knowing what is the contribution of each feature to the prediction.
Using Gradient Boosted Trees models allows us to use SHAP for model explainability and hence discover highly correlated bacteria species for each blood test.
For the sake of data privacy, species names were removed from the images. The graph below shows we found some very interesting species for predicting Fasting Triglycerides. Some of the species, when found in high abundances, are correlated to high levels of triglycerides while other bacteria species share the opposite correlation (high bacteria abundances correlate to low levels of triglycerides)

Then, by looking at each “interesting” bacteria with high correlation (and SHAP value) to one of the blood tests, and plotting the abundance histogram of the entire population, we can compute a risk-score for each person, based on the percentile to which he belongs to.

For example, the bacteria in the graph above has a positive correlation to fasting triglycerides (we found this using SHAP values). I.E. high abundance of this bacteria is correlated to high fasting triglycerides values (which obviously has negative effect on our health) — thus, the lower your values would be, the less risk you have.
Metagenomic Taxonomy Features
Working with DNA sequences reveals many challenges in the process of transforming them into meaningful learnable features. One common bioinformatic tool that we utilize for such process is MetaPhlAn.
MetaPhlAn (Metagenomic Phylogenetic Analysis) is a computational tool for profiling the composition of microbial communities from metagenomic shotgun sequencing data.
The idea behind mapping DNA sequences into bacteria composition can be explained with the following diagram
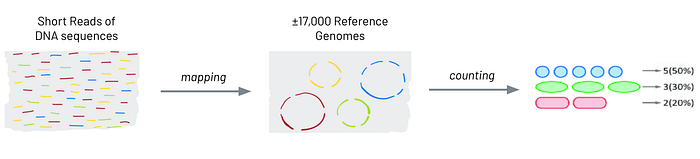
The first step is to generate millions of DNA short reads, each one of length 100 nucleotides, from each stool sample. For each user, all reads are saved into a single FASTQ file, where each row represents a single read.
The second step is mapping each read to a single reference genome. In general, each reference gnome represents a single bacteria, and we use MetaPhlAn as our mapping index. Many errors might occur in this step but we won’t go into them here.
Lastly, counting and normalizing the number of reads we have for each bacteria is what gives us our final features to be used in our machine learning models.
Epilogue
The gut microbiome has been gaining momentum in recent years and there has been a very high increase in interest in it. We are witnessing great research being done in the microbiome domain which links it to various medical conditions — and it is no longer just diabetes.
Pharma and biotechnology companies have already identified the medical potential inherent in each person’s unique microbiome, and it is likely that in the coming years we will see more personalized medicine based on the microbiome.
At DayTwo, we leverage our science with AI capabilities to make sure we uncover many of the hidden information yet to be found in the gut microbiome.
Exciting times are ahead of us.
If you’d like to hear more about the latest and greatest we’ve been working on, don’t hesitate to contact me.
